back to Research
Arteriovenous Malformations and HHT
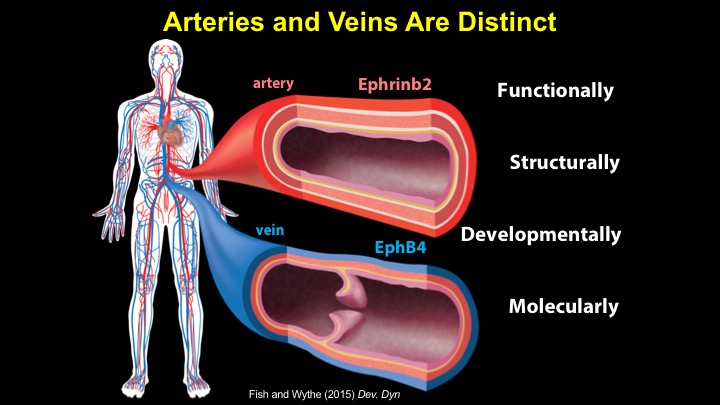
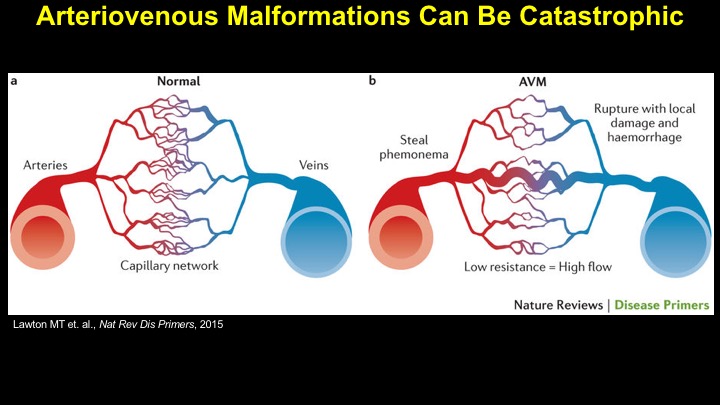
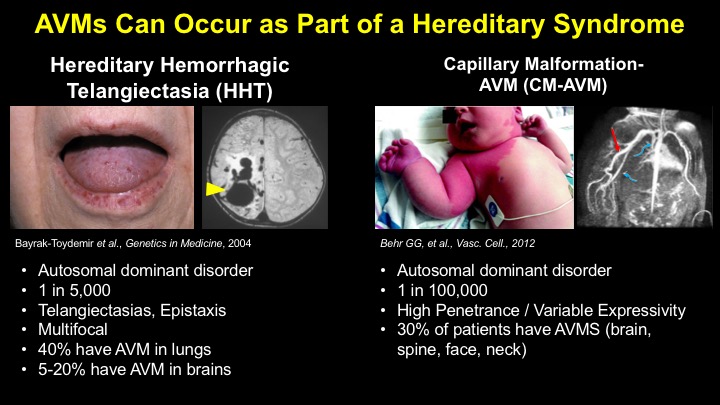
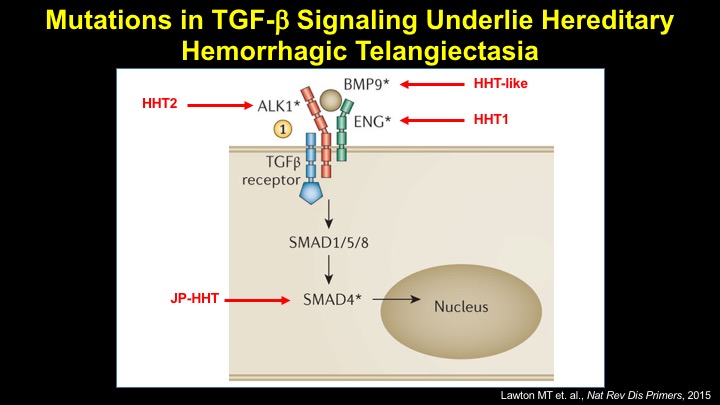
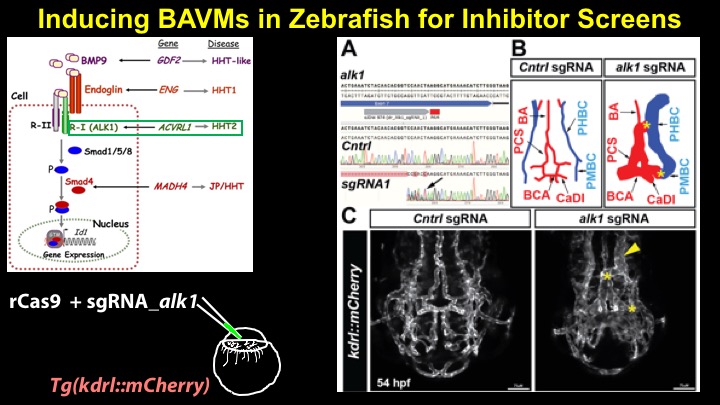
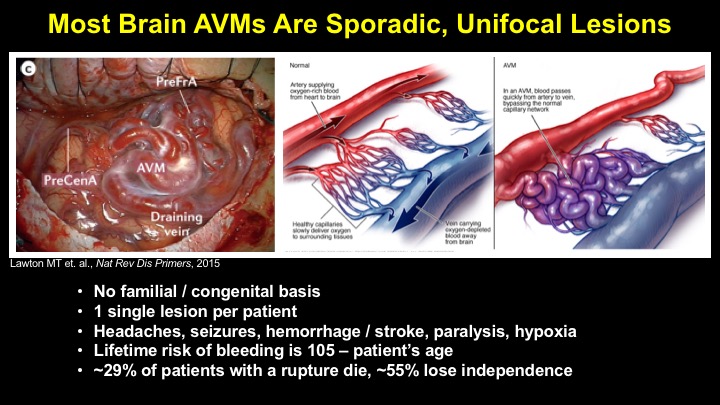
For a more detailed discussion of arteriovenous malformations (AVMs) and the molecular determination of AV identity, we refer interested readers to our recent review (Fish and Wythe, 2015). Briefly, our laboratory is interested in the mechanisms that initially determine arterial-venous identity within the vertebrate embryo during vasculogenesis, and how these signals are altered in pathological settings where arterial-venous boundaries are lost (e.g. AVMs). What we do know, historically, has largely been gained from studying the genes underlying HHT (Hereditary Hemorrhagic Telangiectasia). Work from several groups identified the genetic lesions in ALK and ENDOGLIN, which encode TGF-b co-receptors, in HHT patients. Subsequently, Dean Li's lab at Utah made the first knockout mouse models of these genes and demonstrated that their loss led to AV patterning defects and embryonic lethality. Since those seminal discoveries, mutations have been identified in other TGF-b pathway players, further confirming a role for this signaling network in determining and maintaining AV identity. However, HHT is a genetic syndrome that features AVMs (one of various defects), and many patients that present with other features (mucosal telangiectasias and nosebleeds) do not display AVMs. Furthermore, one of the most catastrophic category of AVMs, brain AVMs (or BAVMs) are more rare than AVMs in other organs in HHT patients. Our recent work (Nikolaev et al., 2018, NEJM), led by Ivan Radovanovic and Jason Fish (at the University of Toronto) and Sergey Nikolaev (formerly at the University of Geneva), found that the more commonly occurring BAVMs, which are unifocal (e.g. one AVM per patient) and non-congenital (e.g. no familial history) feature mutations in KRAS within the endothelium, as well as activation of the downstream MEK/ERK kinase cascade. Future work will determine the effect of these mutations in vivo, as well as the commonality of this pathway's dysregulation in BAVMs.